



在Blender中可视化.xyz结构
无论是在纸上,在海报上还是在演讲中,我们研究的结构的视觉表示都是至关重要的。必须在影响和清晰度之间达成良好的平衡。视情况而定,必须在以清晰简洁的方式传达原子构型的表示形式之间做出选择。还有一个更吸引人,更醒目的视觉效果 但是在这两种情况下,最终的数字都应该尽可能令人赏心悦目。
有许多工具可以操纵,检查和渲染分子和原子结构。但是,与大多数工具一样,最好的工具是高度专业化的,可能无法单枪匹马地以理想的质量实现上述所有目标。今天,我们将使用Python在Blender中渲染原子的配置。
出于一个让我逃避的原因,许多科学家无视与科学传播有关的任何事情。有些人甚至对完全避免在视觉上或其他方面交流其科学的任何尝试感到有点反感。好像花费太多的精力使他们的图表易于理解,他们的人物有吸引力,他们的写作引人入胜以及他们的文章易于阅读,可能会以某种方式削弱或稀释基础科学。为什么是这样,这超出了我。但是,如果您参加了几次会议或阅读了几篇论文,您将会看到类似以下的数字:
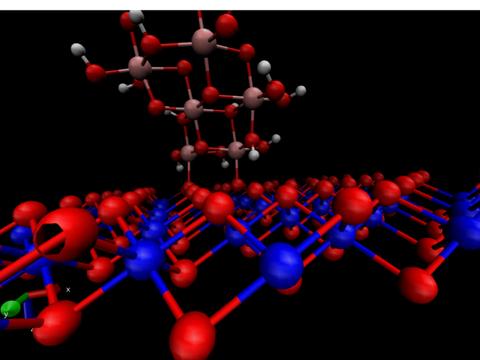
凹凸不平的球体,低劣的塑料外观,默认的背景和照明,无用的轴周围漂浮,甚至最前沿的还有一些剪裁文物。一言以蔽之:丑陋。
以上试图表示的是在顶部具有羟基氧化铁纳米颗粒的氧化镍层。并准许使用适当的标题提供该信息。但是,渲染效果不佳-很难看。
我使用了VMD(可视分子动力学)来绘制图形。虽然我非常喜欢使用此应用程序,但在这里我使用了默认设置并直接呈现了用户的视图,这说明了最终结果的质量很低。但是,在实践中,我会尝试设置,灯光,颜色和材料,以使结构清晰,希望也能使旁观者满意:
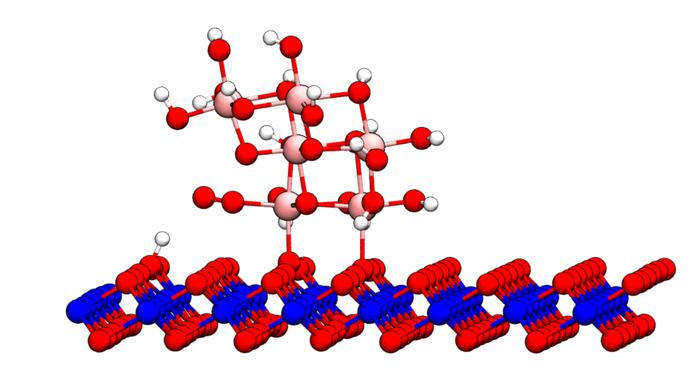
在正交投影中查看的结构相同。明亮的背景,鲜艳的色彩和原子的轮廓使系统清晰易懂。
就个人而言,我更喜欢上述正交投影,因为我发现它比周期性透视图更好地传达周期性结构。但是,假设我们需要一个简短的会议演示图,或者将其作为出版物中的TOC图形来使用。这将在清晰度和视觉效果之间取得平衡,从而有利于后者。然后的任务是创建我们在有限的时间内允许的最具吸引力的表示形式。
如前所述,我们只能使用单个应用程序或程序。尽管VMD在检查结构和轨迹方面非常出色,但使用专用的渲染程序来渲染图形比我们要好得多。CueBlender,一个免费且开放的3D渲染软件。
将Blender和Python连接
我们必须打开Blender并在其中使用Python脚本,而不是将某些Blender包导入Python并使用它在Python中渲染东西。有点相反,但这就是事实。
通过控制台/命令提示符启动Blender。这很重要,因为任何Python错误都只会在控制台中显示。打开一个新的Blender项目。通过单击主窗口的右上角并将其拖动到左侧来拆分视图。然后,单击新面板左下方的小立方体图标,然后选择“文本编辑器”。您应该看到一个空白的灰色文档:
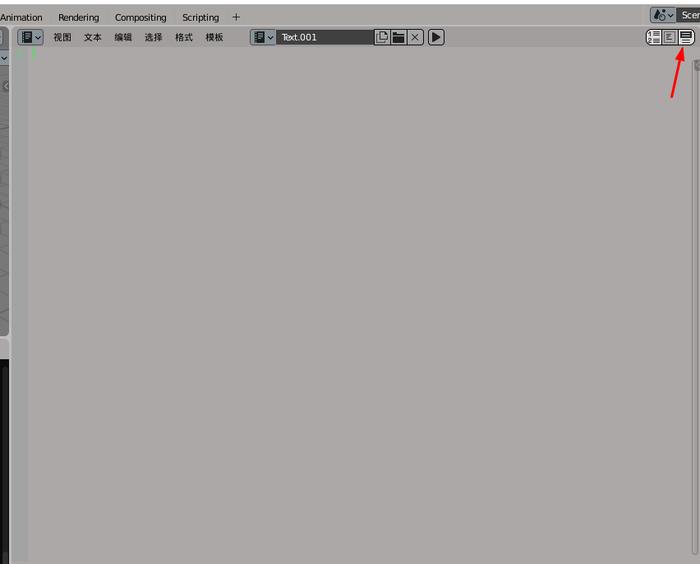
您可以通过按新创建的面板底部同一水平条上的三个图标的最右边来打开语法突出显示。
接下来是import bpy,该软件包使您可以与Blender进行交互。如果您尚未注意到,将光标悬停在几乎所有内容上方,将会在中显示适当的Python函数,方法或模块bpy。现在,用alt + p!运行(空)脚本!
绘制.xyz结构
正如我们在径向分布函数中讨论的那样,.xyz文件格式是原子配置的人类可读表示。第一行包含原子数。第二个保留给评论,将被忽略;其余各行分别包含元素的符号和原子的笛卡尔坐标(以埃为单位)。我们将编写一个脚本,该脚本读取坐标并绘制一个以每个原子的位置为中心的球体。我们还将找到并绘制附近原子之间的键。
importbpy
importnumpy as np
sizes={'Ni':0.7,'Fe':0.7,'O':0.5,'H':0.2}
colors={'Ni': (0.0,0.0,0.7),'Fe': (0.4,0.4,0.7),
'O': (1.0,0.0,0.0),'H': (1.0,1.0,1.0),
'bond': (0.05,0.05,0.05) }
forkeyincolors.keys():
bpy.data.materials.new(name=key)
bpy.data.materials[key].diffuse_color=colors[key]
bpy.data.materials[key].specular_intensity=0.0
defdistance(a, b):
returnnp.sqrt(np.dot(a-b, a-b))
defnormalize(a):
returnnp.array(a)/np.sqrt(np.dot(a, a))
我们首先创建两个字典colors和sizes,分别定义代表每个元素的球的颜色(以RGB为单位)和大小(以埃为单位)。按照惯例,我选择红色代表氧气,白色代表氢气。镍将是深蓝色,铁将是一种钢灰色。接下来,我们为每个元素创建具有适当颜色的材料。我将镜面反射系数设为零,因为我的目标是使外观更具卡通色彩,沉闷,非光泽。在此过程中,我们定义了两个小函数:一个用于计算numpy数组表示的两个点的距离;另一个用于计算由数组表示的两个点的距离。另一个以相同的格式归一化向量。
接下来,我们将创建一个类来表示我们的结构。唯一的初始化参数将是我们要可视化的.xyz文件的路径:
classStructure:
def__init__(self, filename):
withopen(filename,'r') as f:
data=f.readlines()
self.n_atoms=int(data[].split()[])
self.atom_list=[line.split()[]forlineindata[2:]]
coords=[]
fori, lineinenumerate(data[2:]):
coords.append([float(value)forvalueinline.split()[1:4]])
self.coordinates=np.array(coords)
self.bonds=set()
...
.xyz文件的解析遵循RDF帖子中列出的相同逻辑。现在我们已经阅读了原子的坐标,我们可以继续确定它们之间的键。我们在这里采用非常基本的,纯粹基于距离的方法。作为用户,我们可以指定要评估结合的元素列表以及这些结合的截止距离。例如,氧和氢之间的键很短,约为一埃。相反,一些金属-金属键的长度可以是两个或什至更大。
键应表示为一组元组,其中包含通过键连接的原子的索引。我们使用一个集合是因为:(a)一旦建立债券,就不会改变;(b)每个债券应是唯一的;(c)保证金的顺序无关紧要。Python集是完成这项工作的理想数据结构。
现在让我们评估债券。如前所述,我们采用两个参数:元素列表和距离截止。然后,我们执行一个双循环,遍历每对原子。如果两个原子都在元素列表中,并且它们的距离都在临界值以下,则将一个键添加到集合中。非常简单:
classStructure:
...
defadd_bonds(self, atom_types, cutoff):
foriinrange(self.n_atoms-1):
position_1=self.coordinates[i]
element_1=self.atom_list[i]
ifnotelement_1inatom_types:
continue
forjinrange(i+1,self.n_atoms):
position_2=self.coordinates[j]
element_2=self.atom_list[j]
ifnotelement_2inatom_types:
continue
dist=distance(position_1, position_2)
ifdist=cutoff:
self.bonds.add((i, j))
...
准备好原子列表和键列表之后,我们现在就可以在Blender中表示它们:
classStructure:
...
defdraw_structure(self):
self.draw_atoms()
self.draw_bonds()
defdraw_atoms(self):
forelement, positioninzip(self.atom_list,self.coordinates):
bpy.ops.mesh.primitive_uv_sphere_add(size=sizes[element], location=position)
bpy.context.active_object.data.materials.append(bpy.data.materials[element])
bpy.ops.object.shade_smooth()
结合在一起。前者很简单-以坐标列表中每个位置为中心绘制一个球体。其半径和颜色(或更确切地说,材料)由相应的元素控制。
债券的发行还涉及一些时间。对于每个键,我们必须绘制一个圆柱体,其高度等于键合的原子之间的距离,并以原子之间的中点为中心,并正确地定向为引导。我们进行如下。首先,我们计算从一个原子指向另一个原子的向量。接下来,我们找到连接两个原子的直线的中点以及它们之间的距离。在最初将每个新圆柱垂直绘制时,我们将需要旋转它。为此,我们计算了垂直方向上的单位向量与较早计算出的键方向上的归一化向量之间的角度(通过标量积)。我们还对这两个向量进行叉积以得到适当的旋转轴。最后,为缸分配各自的“粘结”材料,
classStructure:
...
defdraw_bonds(self):
foratom_1, atom_2inself.bonds:
pos_1=self.coordinates[atom_1]
pos_2=self.coordinates[atom_2]
difference=pos_2-pos_1
center=(pos_2+pos_1)/2.0
magnitude=distance(pos_1, pos_2)
bond_direction=normalize(difference)
vertical=np.array((0.0,0.0,1.0))
rotation_axis=np.cross(bond_direction, vertical)
angle=-np.arccos(np.dot(bond_direction, vertical))
bpy.ops.mesh.primitive_cylinder_add(radius=0.1,
depth=magnitude,
location=center)
bpy.context.active_object.data.materials.append(bpy.data.materials['bond'])
bpy.ops.object.shade_smooth()
bpy.ops.transform.rotate(value=angle, axis=rotation_axis)
...
做完了 现在,我们可以继续阅读.xyz文件,找到所有相关的键,并在Blender中绘制结构。运行脚本alt + p时应立即导致在相邻窗口中实现原子排列:
NiO2_FeO2=Structure('NiO2_FeO2.xyz')
NiO2_FeO2.add_bonds(('Ni','O','Fe'),2.4)
NiO2_FeO2.add_bonds(('O','H'),1.5)
NiO2_FeO2.draw_structure()
使它看起来不错
我不是Blender专家-机会是,您可以制作出更好的结构效果图。在这里,我将仅描述我为获得卡通效果所做的工作。如果您想拥有闪亮的反射原子,或者想要添加灯光,改变视角或体验花哨的后期处理效果,请成为我的客人。实际上,这就是我想要实现的目标—一旦将结构导入Blender,您就可以自由地做自己想做的事情!
对于所有材质,我都从Lambertian阴影切换为菲涅耳算法。在世界设置中,我玩转背景色,产生了微妙的灰色渐变。接下来,我增加了环境光的强度以柔化阴影。最后,我进行了边缘检测以将原子的轮廓涂成黑色。等等:
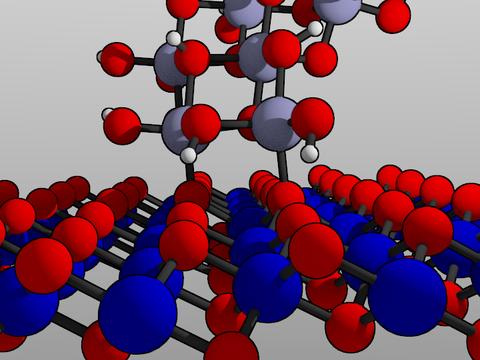
直到下一次!
为了方便起见,我在这里重现整个脚本:
importbpy
importnumpy as np
sizes={'Ni':0.7,'Fe':0.7,'O':0.5,'H':0.2}
colors={'Ni': (0.0,0.0,0.7),'Fe': (0.4,0.4,0.7),
'O': (1.0,0.0,0.0),'H': (1.0,1.0,1.0),
'bond': (0.05,0.05,0.05) }
forkeyincolors.keys():
bpy.data.materials.new(name=key)
bpy.data.materials[key].diffuse_color=colors[key]
bpy.data.materials[key].specular_intensity=0.1
defdistance(a, b):
returnnp.sqrt(np.dot(a-b, a-b))
defnormalize(a):
returnnp.array(a)/np.sqrt(np.dot(a, a))
classStructure:
def__init__(self, filename):
withopen(filename,'r') as f:
data=f.readlines()
self.n_atoms=int(data[].split()[])
self.atom_list=[line.split()[]forlineindata[2:]]
coordinates=[]
fori, lineinenumerate(data[2:]):
coordinates.append([float(value)forvalueinline.split()[1:4]])
self.coordinates=np.array(coordinates)
self.bonds=set()
defadd_bonds(self, atom_types, cutoff):
foriinrange(self.n_atoms-1):
position_1=self.coordinates[i]
element_1=self.atom_list[i]
ifnotelement_1inatom_types:
continue
forjinrange(i+1,self.n_atoms):
position_2=self.coordinates[j]
element_2=self.atom_list[j]
ifnotelement_2inatom_types:
continue
dist=distance(position_1, position_2)
ifdist=cutoff:
self.bonds.add((i, j))
defdraw_bonds(self):
foratom_1, atom_2inself.bonds:
pos_1=self.coordinates[atom_1]
pos_2=self.coordinates[atom_2]
difference=pos_2-pos_1
center=(pos_2+pos_1)/2.0
magnitude=distance(pos_1, pos_2)
bond_direction=normalize(difference)
vertical=np.array((0.0,0.0,1.0))
rotation_axis=np.cross(bond_direction, vertical)
angle=-np.arccos(np.dot(bond_direction, vertical))
bpy.ops.mesh.primitive_cylinder_add(radius=0.1,
depth=magnitude,
location=center)
bpy.context.active_object.data.materials.append(bpy.data.materials['bond'])
bpy.ops.object.shade_smooth()
bpy.ops.transform.rotate(value=angle, axis=rotation_axis)
defdraw_atoms(self):
forelement, positioninzip(self.atom_list,self.coordinates):
bpy.ops.mesh.primitive_uv_sphere_add(size=sizes[element], location=position)
bpy.context.active_object.data.materials.append(bpy.data.materials[element])
bpy.ops.object.shade_smooth()
defdraw_structure(self):
self.draw_atoms()
self.draw_bonds()
NiO2_FeO2=Structure('NiO2_FeO2.xyz')
NiO2_FeO2.add_bonds(('Ni','O','Fe'),2.4)
NiO2_FeO2.add_bonds(('O','H'),1.5)
NiO2_FeO2.draw_structure()
发布于:浙江


举报成功